Einführung in die Multiphotonen-Fluoreszenzmikroskopie
Die Multiphotonen-Fluoreszenzmikroskopie ist eine leistungsfähige Methode in der Forschung, die moderne optische Techniken der Laser-Raster-Mikroskopie mit der langwelligen Multiphotonen-Fluoreszenzanregung kombiniert, um hochauflösende, dreidimensionale Bilder von mit hochspezifischen Fluorophoren markierten Proben aufzunehmen.

Abbildung 1: Konfiguration eines Fluoreszenzmikroskops mit Multiphotonen-Anregung
Die Methode ist besonders für Zellbiologen interessant, die dynamische Prozesse in lebenden Zellen und Geweben untersuchen wollen, ohne die Proben erheblich – und oft tödlich – zu schädigen. Auch wenn die klassische Weitfeld-Fluoreszenzmikroskopie bei biochemischen Vorgängen in lebenden Systemen oft eine Auflösung im Submikrometerbereich erreicht, sind die Empfindlichkeit und die räumliche Auflösung dieser Technik durch das Hintergrundrauschen begrenzt, das durch sekundäre Fluoreszenz oberhalb und unterhalb der Fokusebene verursacht wird.
Bei der Multiphotonenmikroskopie erfolgt die Anregung nur im Brennpunkt eines beugungsbegrenzten Mikroskops, so dass sich dicke biologische Proben für eine dreidimensionale Auflösung optisch in Schnitte aufteilen lassen. Die einzelnen optischen Schnitte werden durch Rasterabtastung der Probe in der x-y-Ebene erfasst. Daraufhin wird ein vollständiges dreidimensionales Bild durch serielle Abtastung der Probe an aufeinanderfolgenden z-Positionen erstellt. Da die Position des Brennpunkts genau bestimmt und kontrolliert werden kann, eignet sich die Multiphotonen-Fluoreszenzmikroskopie zur Untersuchung ausgewählter Bereiche unter der Probenoberfläche. Die hochgradig lokalisierte Anregungsenergie minimiert das Photobleaching der an der Probe haftenden Fluorophore und verringert Lichtschäden, so dass sich die Lebensfähigkeit der Zellen und die Dauer der Experimente zur Untersuchung der Eigenschaften lebender Zellen verlängern. Darüber hinaus ermöglicht die Verwendung von Anregungswellenlängen im nahen Infrarotspektrum ein tieferes Eindringen in biologische Materialien und reduziert die starke Lichtstreuung, die bei kürzeren Wellenlängen zu beobachten ist. Dank dieser Vorteile können Experimente an dicken Proben von Lebendgewebe durchgeführt werden, die mit anderen Mikroskopietechniken nur schwer oder gar nicht abgebildet werden können, z. B. an Gehirnschnitten oder Gehirnen lebender Tiere in vivo sowie an sich entwickelnden Embryonen.
Abbildung 1 zeigt eine typische Konfiguration, wie sie in Experimenten zur Multiphotonen-Fluoreszenzmikroskopie verwendet wird. Das Mikroskop ist ein aufrechtes Gerät zur Beobachtung von lebendem Gewebe kleiner Versuchstiere in vivo. An der Rückseite des Mikroskopstativs befindet sich ein modengekoppeltes gepulstes Lasersystem mit einem Titan-Saphir, das aufgrund der hohen Spitzenintensität und der geringen Durchschnittsleistung eine der bevorzugten Quellen für die Multiphotonen-Anregung ist. Ein gefiltertes Photomultiplier-Detektionssystem befindet sich gerade nahe genug am Objektivrevolver des Mikroskops, um die vom Objektiv effektiv erfasste gestreute Fluoreszenz zu erkennen. Die mit dem Mikroskop erfassten digitalen Bilder werden von einer Computer-Workstation verarbeitet und analysiert, die aus optischen Schnitten dreidimensionale Rekonstruktionen erstellen kann.
Nachteil der herkömmlichen Weitfeld-Fluoreszenzmikroskopie ist die Sekundärfluoreszenz außerhalb des Fokusbereichs, die Fingerspitzengefühl erfordert und zu einem starken Hintergrundrauschen beiträgt, so dass oft wichtige Details der Probe verdeckt bleiben. Die konfokale Mikroskopie umgeht dieses Problem weitgehend, indem sie die nicht im Fokus liegende Hintergrundfluoreszenz durch den Einsatz von Lochblenden ausschließt und dünne (weniger als ein Mikrometer dicke), scharfe optische Schnitte aus dem Inneren dicker Proben erzeugt. Die Einführung der Multiphotonen-Fluoreszenzmikroskopie ist durch selektive Anregung sowie eine breitere Palette von Detektionsmöglichkeiten eine Alternative zur konfokalen Mikroskopie. Im Gegensatz zu herkömmlichen konfokalen Mikroskopen benötigt das in Abbildung 1 dargestellte Mikroskop keine Lochblende in der Nähe des Detektors zur dreidimensionalen Unterscheidung, was die Effizienz der emittierten Fluoreszenzsignale deutlich erhöht. In der Vergangenheit schränkten die hohen Kosten und die Komplexität der gepulsten Lasersysteme für die Multiphotonen-Anregung den Einsatz dieser Technik ein. Dank schlüsselfertiger Laser- und kommerzieller Multiphotonen-Systeme ist die Multiphotonen-Fluoreszenzmikroskopie heute die bevorzugte Methode für viele Untersuchungen.
Zwei-Photonen- und Drei-Photonen-Anregung
Die Grundprinzipien der Multiphotonen-Anregung wurden erstmals von Maria Göppert-Mayer, Ph.D., im Rahmen der Forschung zu ihrer Dissertation vor über 70 Jahren beschrieben, aber die Hypothese konnte erst mit der Erfindung der gepulsten Rubinlaser etwa 30 Jahre später bestätigt werden. Bei hohen Photonendichten können zwei Photonen gleichzeitig absorbiert werden (vermittelt durch einen virtuellen Zustand), deren Energien zusammen den elektronischen Übergang eines Fluorophors in den angeregten Zustand bewirken. Da die Energie eines Photons umgekehrt proportional zu seiner Wellenlänge ist, sollten die beiden Photonen etwa die doppelte Wellenlänge haben, die für die Ein-Photonen-Anregung erforderlich ist. Beispielsweise können zwei Photonen mit einer Wellenlänge von 640 Nanometern (rotes Licht) zusammen ein Ultraviolett absorbierender Fluorophor im 320-Nanometer-Bereich (ultraviolett) anregen und eine sekundäre Fluoreszenzemission mit längerer Wellenlänge (blau oder grün) auslösen. Durch diese einzigartige Anwendung lassen sich somit vorteilhaft längere Wellenlängen bis in den Infrarotbereich hinein zur Anregung von Chromophoren mit einem einzigen Quantenereignis nutzen, die dann Sekundärstrahlung mit niedrigeren Wellenlängen aussenden.
Da für jedes Anregungsereignis zwei Photonen benötigt werden, ist eine Ratenkonstante erforderlich, die vom Quadrat der Anregungsintensität abhängt. Auch wenn die Photonen für eine Multiphotonen-Anregung nicht die gleiche Wellenlänge haben müssen, besitzen die meisten Versuchssysteme nur eine einzige Laserquelle, so dass die beiden Photonen in der Regel zu einer bestimmten Population mit enger Wellenlängenverteilung gehören. Anders als bei der Ein-Photonen-Absorption hängt die Wahrscheinlichkeit, dass ein bestimmter Fluorophor gleichzeitig zwei Photonen absorbiert, von der räumlichen und zeitlichen Überlappung der einfallenden Photonen ab. Nach Berechnungen, bei denen angenommen wird, dass jeder Fluorophor dem gleichen Laserquerschnitt ausgesetzt ist, müssen die Photonen mit einem zeitlichen Abstand von 10(-18) Sekunden (einer Attosekunde) eintreffen. Die Zeitskala dieser Überlappungsperiode entspricht der Lebensdauer (10(-17) Sekunden oder 0,01 Femtosekunde) des virtuellen Zwischenzustands.
Bei der Multiphotonen-Fluoreszenz sind hohe Photonendichten erforderlich, um eine ausreichende Anregung der Fluorophore zu gewährleisten. Tatsächlich muss die Photonenkonzentration etwa eine Million Mal so hoch sein wie für eine entsprechende Anzahl von Ein-Photonen-Absorptionen. Dies wird mit leistungsstarken, modengekoppelten, gepulsten Lasern erreicht, die während der Impulsspitzen eine beträchtliche Leistung erzeugen, deren durchschnittliche Leistung jedoch so gering ist, dass die Probe nicht beschädigt wird. Kurze, aber intensive Laserimpulse erhöhen die durchschnittliche Zwei-Photonen-Absorptionswahrscheinlichkeit für einen bestimmten Fluorophor bei konstanter durchschnittlicher Laserleistung. Die Minimierung der durchschnittlichen Anregungsleistung verringert die Ein-Photonen-Absorption, die während der Anregung auch in der Probe auftritt. Bei Fluoreszenzexperimenten sind es die Ein-Photonen-Anregungen, die einen Großteil der Erwärmung und einen Teil der Lichtschäden verursachen.
Typische gepulste Laserkonfigurationen für Multiphotonen-Fluoreszenzexperimente haben kurze Arbeitszyklen von etwa 100 Femtosekunden (10 e(-13) Sekunden) mit einer Wiederholrate von 80 bis 100 Megahertz. Dieses Verfahren ermöglicht eine zufriedenstellende Bildaufnahme, ohne dass die Probe einer übermäßigen Wärmeentwicklung und Lichtschäden ausgesetzt wird. Die Zeitdauer jedes Impulses wird zwar oft als „ultrakurz“ bezeichnet, ist aber immer noch vier bis fünf Größenordnungen länger als die Reaktionszeit für die Zwei-Photonen-Absorption. Die Population der Singulett-Zustände in Chromophoren, die durch einen Zwei-Photonen-Impuls angeregt werden, ist identisch mit der Population bei herkömmlicher Weitfeld- oder konfokaler Fluoreszenzmikroskopie. Daher ist die sekundäre Fluoreszenzemission nach Zwei-Photonen-Anregung nicht von der in Ein-Photonen-Experimenten beobachteten zu unterscheiden. Ein Fluorophor, z. B. Rhodamin, emittiert den gleichen breiten Wellenlängenbereich als Sekundärfluoreszenz, unabhängig davon, ob er durch ein oder zwei Photonen angeregt wurde.
Multiphotonen-Anregung - Jablonski-Schema
Erfahren Sie, wie Fluoreszenzanregungen in der Ein-Photonen-, Zwei-Photonen- und Drei-Photonen-Mikroskopie unter Verwendung des klassischen Jablonski-Schemas ablaufen.
Die Drei-Photonen-Anregung ist ein nichtlineares optisches Absorptionsereignis, das in ähnlicher Weise wie die Zwei-Photonen-Anregung auftreten kann. Der Unterschied besteht darin, dass drei Photonen gleichzeitig eine Wechselwirkung mit dem Fluorophor haben müssen, um einen Übergang in den angeregten Singulett-Zustand zu bewirken. Vorteil der Drei-Photonen-Anregung ist, dass für eine erfolgreiche Absorption nur eine zehnmal höhere Photonenkonzentration als bei der Zwei-Photonen-Absorption erforderlich ist, was diese Technik für einige Experimente attraktiv macht. Die Drei-Photonen-Anregung kann die Auflösung in der z-Achse noch stärker verbessern als die Zwei-Photonen-Absorption. Dies ist auf den geringeren Wirkungsquerschnitt für die Fluorophoranregung zurückzuführen, der dadurch entsteht, dass eine gleichzeitige Wechselwirkung mit drei einzelnen Photonen erforderlich ist. In der Praxis kann ein Laser, der infrarotes Licht mit einer auf 1050 Nanometer zentrierten Wellenlängenverteilung aussendet, einen Fluorophor anregen, der im ultravioletten Bereich absorbiert (etwa 350 Nanometer, ein Drittel der Anregungswellenlänge). Derselbe Laser kann gleichzeitig einen anderen Fluorophor bei der halben Wellenlänge (525 Nanometer) anregen, eine nützliche Kombination für biologische Experimente mit zwei Markierungen.
Durch Verwendung kürzerer Wellenlängen im nahen Infrarotspektrum (bis 720 Nanometer) erweitert die Drei-Photonen-Fluoreszenz den nutzbaren Fluoreszenz-Bildgebungsbereich bis ins tiefe Ultraviolettspektrum. Laserwellenlängen im Bereich von 900 bis 700 Nanometern regen Fluorophore an, die im Bereich von 240 bis 300 Nanometern absorbieren, was mit herkömmlichen Mikroskopoptiken praktisch unmöglich darstellbar ist. Das zur Herstellung von Fluoreszenzobjektiven verwendete Glas hat eine sehr geringe Transmission für Wellenlängen unter 300 Nanometern, längerwellige Infrarotlaserstrahlung dagegen lässt es ohne Weiteres durch und kann so eine Drei-Photonen-Anregung erzeugen.
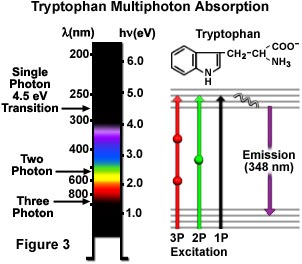
In Abbildung 3 sind Einzel-, Doppel- und Dreifach-Photonen-Anregungen einer häufigen aromatischen Aminosäure (Tryptophan) schematisch dargestellt. Ein Ein-Photonen-Übergang mit 4,5 Elektronenvolt regt Tryptophan bei 280 Nanometern an und führt zu einer Sekundärfluoreszenz bei 348 Nanometern im ultravioletten Bereich. Die Anregung durch den Zwei-Photonen-Mechanismus erfolgt mit grünlich-gelbem Licht, zentriert auf 580 Nanometer, die Drei-Photonen-Anregung der Aminosäure dagegen mit 840 Nanometern im nahen Infrarotbereich. Die Übergänge werden in einem Jablonski-Schema dargestellt (Abbildung 3), wobei der virtuelle Zustand bei Zwei-Photonen-Anregung durch eine Kugel und bei Drei-Photonen-Anregung durch zwei Kugeln dargestellt wird. Tryptophan besitzt eine viel stärkere Fluoreszenz mit einer höheren Quantenausbeute als die anderen aromatischen Aminosäuren und ist in den meisten Proteinen nur in geringen Mengen vorhanden. Diese Eigenschaften machen die Multiphotonenmikroskopie zu einem hervorragenden Instrument für Untersuchungen, die die Autofluoreszenz von Tryptophanresten nutzen. Es sind sogar nichtlineare Phänomene höherer Ordnung möglich, beispielsweise eine Vier-Photonen-Anregung, die in der biologischen Forschung jedoch noch nicht genutzt wurden.
Zwei-Photonen-Fluoreszenzmikroskopie
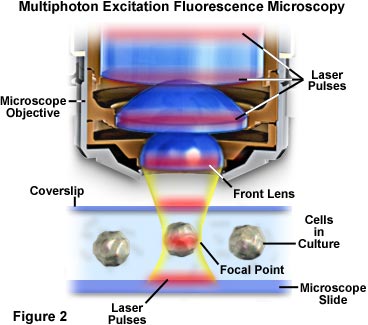
Bei der Multiphotonenmikroskopie wird die Anregung auf den Bereich in unmittelbarer Nähe des Brennpunkts beschränkt, weil dort die Photonendichte am höchsten ist. Dieser Vorteil ergibt sich aus dem grundlegenden physikalischen Prinzip, dass die Zwei-Photonen-Absorption eines Fluorophors vom Quadrat der Anregungsintensität abhängt. Wenn Photonen aus einer gepulsten Laserquelle durch ein Objektiv mit hoher numerischer Apertur fokussiert werden, erhöht sich die Anzahl der Photonen und damit die Wahrscheinlichkeit, dass zwei oder mehr davon gleichzeitig eine Wechselwirkung mit einem einzelnen Fluorophor haben. Die Konzentration der Photonen im Brennpunkt des Mikroskops ist für die Mehrphotonenabsorption entscheidend, weil dies der einzige Bereich ist, in dem eine nennenswerte Anregung stattfindet. Das Konzept ist in den Abbildungen 2 und 4 dargestellt, welche die Multiphotonen-Anregung auf makroskopischer bzw. mikroskopischer Ebene veranschaulichen. Abbildung 2 zeigt die vergrößerte Ansicht eines Mikroskopobjektivs in der Position, in der es kultivierte Zellen auf einem Objektträger mit Deckglas abbildet. Rote Laserimpulse durchqueren die Längsachse des Objektivs und werden auf die Zelle im mittleren Teil der Abbildung fokussiert und konzentriert.
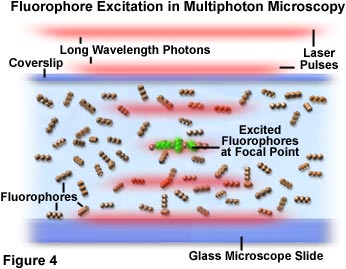
In Abbildung 4 ist die Ansammlung von Photonen und die Wechselwirkung mit Fluorophoren im Brennpunkt des Mikroskops zu erkennen. Wenn die Impulse des roten Laserlichts die Probe mit den Fluorophoren (dargestellt als lineares Kugeltriplett) durchdringen, erhöht sich die Wahrscheinlichkeit der Anregung, wenn die Impulse den Brennpunkt des Objektivs erreichen. Einzelne Photonen werden als Anhäufung dargestellt, die in diffuse rote Linien aufgeteilt ist, die die Grenzen der Laserimpulse darstellen. Eine kleine Gruppe von Fluorophormolekülen in der Mitte des Fokusbereichs in Abbildung 4 wurde durch die gleichzeitige Absorption von zwei Photonen angeregt und erzeugt eine grüne Sekundärfluoreszenz. Die Wahrscheinlichkeit, dass Chromophore außerhalb der Fokusebene zwei Photonen absorbieren, ist nahezu null, da die Photonendichte in diesem Bereich nicht hoch genug ist.
Das Phänomen der Zwei-Photonen-Anregung ist nicht nur durch die räumliche Nähe der Fluorophore im Brennpunkt des Mikroskops möglich, sondern auch durch die zeitliche Überlappung der in den aufeinanderfolgenden Laserimpulsen enthaltenen Photonen. Wie bereits erwähnt, verhält sich die Anregungsenergie bei der Zwei-Photonen-Absorption proportional zum Quadrat der von der Laserquelle erzeugten Photonenintensität. Die Intensität des gepulsten Laserstrahls nimmt im Quadrat des Abstands von der Fokusebene ab, so dass die Anregungswahrscheinlichkeit eines Fluorophors in der Nähe des Fokusbereichs mit der vierten Potenz des Fluorophorabstands von der Fokusebene sinkt. Die Maße des gepulsten Laser-Beleuchtungskegels werden durch die numerische Apertur des Objektivs bestimmt. Somit ist die Abnahme der Strahlintensität vom Brennpunkt proportional zum Durchmesser des Anregungslichtkegels im Quadrat. Da sich der Beleuchtungskegel oberhalb und unterhalb des Brennpunkts erweitert, nimmt die Wahrscheinlichkeit der Fluorophoranregung mit der vierten Potenz des Kegeldurchmessers ab. Aus diesem Grund beschränkt sich die Anregung der Fluorophore auf die unmittelbare Umgebung des Brennpunkts, der nur einen sehr dünnen optischen Schnitt der gesamten Probe darstellt.
Ereignisse im Erregungsbereich
Untersuchung der Vorgänge im Brennpunkt des Mikroskops während der Probenanregung mit langwelliger Laserbeleuchtung im sichtbaren Spektrum und nahen Infrarotspektrum.
Die Dauer von Laserimpulsen liegt typischerweise zwischen etwa 100 Femtosekunden und 1 Pikosekunde (10 e(-13) bis 10 e(-12) Sekunde) und gilt makroskopisch gesehen als ultrakurz. Gemessen an der Zeitskala für Photonen-Absorptionsereignisse (etwa ein Tausendstel einer Femtosekunde) sind die Impulse jedoch tatsächlich recht lang. Dadurch wird die Sättigung der Fluorophore begrenzt und die Moleküle haben ausreichend Zeit, zwischen den Impulsen in den Grundzustand zurückzukehren, bevor eine weitere Anregung erfolgt. Die Impulswiederholraten liegen zwischen 80 und 120 Megahertz (MHz), was eine hohe momentane Anregungs-Spitzenleistung mit einer anschließenden Verweildauer von durchschnittlich 10 Nanosekunden ermöglicht. Da die Fluoreszenzlebensdauer eines typischen Fluorophors nur einige Nanosekunden beträgt, hat die Population der angeregten Moleküle zwischen den Impulsen viel Zeit zur Erholung. Das relativ kurze Tastverhältnis (die Impulsdauer geteilt durch die Zeit zwischen den Impulsen) begrenzt die durchschnittliche Eingangslaserleistung auf einen Wert, der nur geringfügig über dem Wert liegt, der routinemäßig für die konfokale Laser-Raster-Mikroskopie verwendet wird.
Die Beschränkung der Zwei-Photonen-Anregung auf den Bereich nahe der Fokalebene ist ein wesentlicher Vorteil der Multiphotonen-Mikroskopie gegenüber der konfokalen Mikroskopie. Bei der konfokalen Mikroskopie wird die Fluoreszenz in der gesamten Probe angeregt und die vom Detektor erfasste Sekundärfluoreszenz durch die konfokale Lochblende auf die Brennebene des Objektivs beschränkt. Dadurch wird das Hintergrundrauschen bzw. die Fluoreszenz anderer Fokusebenen reduziert, die die Daten durch Hintergrundrauschen überlagern. Im Gegensatz dazu erzeugt die Multiphotonenmikroskopie die Fluoreszenzanregung (und anschließend die Fluoreszenzemission) nur in der Fokusebene, so dass sowohl das Hintergrundrauschen als auch die Notwendigkeit einer konfokalen Lochblende entfallen. Der deutliche Unterschied zwischen den Anregungsmodi in der konfokalen und der Multiphotonenmikroskopie ist in Abbildung 5 zu erkennen, die einen Überblick über die Photobleachingprofile jeder Technik gibt.
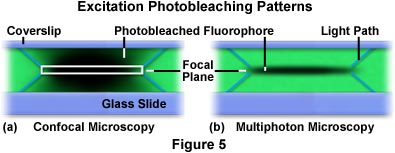
In Abbildung 5 sind die x-z-Muster für Photobleaching dargestellt, die beim wiederholten Abtasten einer x-y-Ebene in einem mit dem Fluorophor Rhodamin (grün) angefärbten formvar-Polymerfilm auftreten. Die Abbildung links (Abbildung 5(a)) zeigt das Profil, das durch Scannen des angefärbten Films mit einem konfokalen Mikroskop erfasst wurde. Das weiße Rechteck in der Mitte des Scans ist die Brennebene, die durch die Lochblende vom Detektor abgebildet wird. Diagonale blaue Linien, die von den oberen und unteren Ecken des Rechtecks ausgehen, stellen den Weg des Anregungslichts durch den Film dar. Wenn das Strahlenraster den Film abtastet, wird der Fluoreszenzfarbstoff angeregt und erzeugt eine Sekundärfluoreszenz. Schließlich kommt es zum Photobleaching, hier durch die dunklen Bereiche im Fokusbereich dargestellt. In dem mit dem konfokalen Mikroskop abgetasteten Film (Abbildung 5(a)) ist die integrierte Anregung über den gesamten Anregungspfad, sowohl oberhalb als auch unterhalb der Brennebene, nahezu gleich. Umgekehrt begrenzt das vom Multiphotonenmikroskop erzeugte x-z-Repetitiv-Scan-Anregungsprofil die Anregung und das Photobleaching auf die Fokusebene (Abbildung 5(b)). Ähnlich wie in Abbildung 5(a) markieren die diagonalen blauen Linien, die von der Brennebene ausgehen, den Weg des Anregungslichts bis zur Brennebene.
Die lokale Anregung der Multiphotonenmikroskopie hat eine Reihe von Vorteilen. Der vielleicht wichtigste Aspekt ist die hohe dreidimensionale Auflösung, die mit dieser Technik erreicht werden kann und mit der eines idealen konfokalen Mikroskops identisch ist. Da an Fluorophoren außerhalb der Fokusebene keine Absorption erfolgt, kann ein größerer Teil des Anregungslichts die Probe durchdringen und die Fokusebene erreichen. Dadurch erhöht sich deutlich die Fähigkeit des fokussierten Strahls, tief in die Probe einzudringen, häufig bis in eine Tiefe, die zwei- bis dreimal so groß ist wie bei der konfokalen Mikroskopie.
Wie bereits erwähnt, sinkt die Wahrscheinlichkeit der Mehrphotonenabsorption außerhalb des Brennpunkts mit der vierten Potenz der Entfernung entlang der optischen Achse (der z-Richtung). Wenn eine gleichmäßige Verteilung von Fluorophoren einer Multiphotonen-Anregung mit einem Objektiv mit hoher numerischer Apertur (1,4) ausgesetzt wird, erfolgen etwa 80 Prozent der Absorption in einem eng definierten Raum, dem sogenannten Fokalvolumen. Die Abmessungen des Fokalvolumens hängen von der numerischen Apertur des Objektivs ab. Für ein typisches Fluoreszenzobjektiv mit großer Apertur im nahen Infrarotspektrum wird dieser Bereich durch ein Ellipsoid mit einem seitlichen Maß von 0,3 Mikrometern Durchmesser und einer axialen Länge von 1 Mikrometer definiert.
Anregungs-Photobleaching-Muster
Vergleich anregungsinduzierter Photobleaching-Muster, die in der Nähe der Fokusregion bei Multiphotonen- und konfokalen Mikroskopiesystemen auftreten.
Die in Abbildung 5(b) gezeigte signifikante Verringerung des Photobleaching (und der damit verbundenen Lichtschäden von Zellen und Geweben) ist bei der Multiphotonenmikroskopie wesentlich geringer als bei der konfokalen Mikroskopie. Photobleaching und Lichtschäden sind die zwei Haupteinschränkungen der Fluoreszenzmikroskopie bei der Untersuchung von lebenden Zellen, Geweben und anderen Organismen. Die Anregung eines Fluorophors führt dazu, dass ein Elektron aus dem Grundzustand in einen angeregten Singulett-Energiezustand übergeht. Während der Schwingungsrelaxation aus dem angeregten Zustand kann es zu einem Intersystemübergang in den Triplett-Zustand kommen, statt zum typischen Rückfall in den Singulett-Grundzustand. Triplett-Zustände sind extrem reaktiv und relativ langlebig, so dass Fluorophore in diesem Zustand Zeit haben, mit lebenden Zellen zu reagieren oder sich durch molekulare Degeneration oder Umstrukturierung in eine nicht fluoreszierende Verbindung zu verwandeln. Darüber hinaus können angeregte Fluorophore im Triplett-Zustand Singulett-Sauerstoff erzeugen, der mit einer Vielzahl von funktionellen Gruppen der benachbarten Biomoleküle reagieren kann. Das Anregungslicht muss die Probe auf dem Weg zum Brennpunkt in allen Brennebenen durchdringen, der größte Teil dieses Lichts breitet sich noch weit hinter den Brennpunkt aus. Dadurch tritt in einer Population von Fluorophoren, die wie bei der Weitfeld- und der konfokalen Mikroskopie im gesamten Strahlengang angeregt wird, ein beträchtliches Photobleaching mit Zell- und Gewebeschäden auf, das mit der Multiphotonen-Technik vermieden werden kann.
Auch wenn die genauen Mechanismen der durch Lichteinwirkung hervorgerufenen Zellschäden nur unzureichend erforscht sind, steht fest, dass eine Verringerung der Lichtschäden die Lebensfähigkeit biologischer Proben, die mit Fluoreszenzmikroskopie untersucht werden, deutlich erhöht. Die Exposition mit langwelligem Licht des sichtbaren und nahe Infrarotspektrums allein scheint die Lebensfähigkeit der Zellen nicht zu beeinträchtigen, so dass es wahrscheinlich ist, dass ein Großteil der mit der Multiphotonenmikroskopie verbundenen Schäden durch die Anregung entsteht und auf die Fokusebene beschränkt ist.
Detektoren für die Multiphotonenmikroskopie
Bei der Multiphotonenmikroskopie stammen die durch Sekundärfluoreszenz emittierten Photonen fast ausschließlich aus der Fokusebene des Objektivs, so dass die Notwendigkeit einer Descanned-Detektion entfällt und flexiblere Detektionsgeometrien möglich sind. Diese Vielseitigkeit im Vergleich zur konfokalen Mikroskopie kann zu einer erheblichen Verbesserung der Fluoreszenzdetektionseffizienz führen. In einem System mit Descanned-Detektion wird das vom Objektiv gesammelte Licht durch die Oberfläche mehrerer Abtastspiegel reflektiert, bevor es durch eine Lochblende zum Detektor gelangt. Die konfokale Lochblende verbessert zwar die Bildauflösung, verschlechtert aber deutlich die Erkennungseffizienz und erfordert eine längere Belichtung der Probe, was die Wahrscheinlichkeit von Lichtschäden und Photobleaching erhöht.
Zur Maximierung der Detektionseffizienz befinden sich die Non-Descanned-Detektoren in der Regel in der Nähe des Objektivs, und der Durchmesser des Lichtwegs muss größer sein, um das streuende Fluoreszenzsignal aus der Tiefe der Probe effektiv zu erfassen. Ein gängiger Detektor für die Multiphotonenmikroskopie ist die Photomultiplier-Röhre (PMT). Beim Einsatz von Galliumarsenidphosphid (GaAsP) PMT-Detektoren können Bilder mit einem hohen Signal-Rausch-Verhältnis selbst bei schwacher Fluoreszenz aufgenommen werden, da die Quanteneffizienz höher ist als bei herkömmlichen Multi-Alkali-PMTs.

Abbildung 6
Auflösung in der Multiphotonenmikroskopie
Die Auflösung in der Multiphotonenmikroskopie ist nicht höher als bei der konfokalen Mikroskopie, und die Nutzung längerer Wellenlängen (rot bis nahes Infrarot, d. h. 700 bis 1200 Nanometer) führt zu einer größeren Punktspreizung für die Multiphotonen-Anregung. Dies bewirkt eine leichte Verringerung der lateralen und axialen Auflösung. Bei einer Anregungswellenlänge von 700 Nanometern und einem Objektiv mit einer numerischen Apertur von 1,3 beträgt die beobachtete laterale Auflösung beispielsweise etwa 0,2 Mikrometer und die axiale Auflösung entsprechend 0,6 Mikrometer. In Verbindung mit der Stokeschen Verschiebung können diese Werte bis zu 30 % über der Auflösung liegen, die mit herkömmlicher konfokaler Mikroskopie unter identischen Bedingungen erreicht wird. In der Praxis kann sich die konfokale Auflösung durch die Maße der Lochblende, chromatische Aberration und unvollkommene Ausrichtung des optischen Systems verschlechtern, so dass sich die Auflösungsunterschiede zwischen konfokaler und Multiphotonenmikroskopie verringern. Aus dieser Diskussion wird offensichtlich, dass Strukturen, die mit einem konfokalen Mikroskop nicht ausreichend aufgelöst werden, auch bei der Multiphotonen-Anregung nicht besser (möglicherweise sogar schlechter) abgebildet werden.
Bei der Aufnahme digitaler Bilder oder der Zählung von Photonen mit dreidimensionaler räumlicher Auflösung ist es wichtig, zwischen der Fluoreszenzemission innerhalb des Fokusvolumens und der Emission aus dem Hintergrund zu unterscheiden. Die Unterscheidung zwischen den beiden Signalen kann instrumentell (mit konfokalen oder Multiphotonen-Mikroskopen) oder durch Dekonvolution eines dreidimensionalen Datensatzes vorgenommen werden. Inwieweit zwischen der Fluoreszenzemission aus der Fokusebene und der Hintergrundfluoreszenz unterschieden werden kann, hängt vom Signal-Hintergrund-Verhältnis (S/B) ab, wobei S die Anzahl oder Intensität der in der Fokusebene erfassten Photonen und B die Anzahl der Photonen aus dem Hintergrund (aus nicht fokussierten Ebenen) ist. Bei der konfokalen Rastermikroskopie werden hohe S/B-Verhältnisse durch die Unterdrückung des Hintergrundsignals mit der konfokalen Lochblende erzielt. Bei der Multiphotonen-Anregung sind die S/B-Verhältnisse von Natur aus groß, da außerhalb der Fokusebene nur eine sehr geringe Anregung stattfindet. Die Auflösungsberechnungen der Multiphotonen- und konfokalen Techniken können verglichen werden, wenn bei den konfokalen Berechnungen eine unendlich kleine Lochblende angesetzt wird. Bei beiden Techniken ist das Signal-Hintergrund-Verhältnis in der Regel mehrere Größenordnungen höher als bei der klassischen Weitfeld-Fluoreszenzmikroskopie.
Ein weiterer wichtiger Aspekt ist, dass die Multiphotonen-Anregung die Verwendung von Fluorophoren mit Absorptionsübergängen im kurzwelligen ultravioletten Bereich ermöglicht. Da die konfokale Mikroskopie Fluorophore unterhalb von etwa 340 Nanometern kaum anregen kann, verwenden die Forscher in der Regel Sonden mit deutlich längeren Wellenlängen und entsprechend geringerer Auflösung. In kritischen Situationen kann die Auflösung in der Multiphotonenmikroskopie durch Beschränkung der Bildgebungswellenlängen und eine konfokale Lochblende bzw. durch die Verwendung eines räumlich aufgelösten Detektionssystems, beispielsweise eines CCD-Photodiodenarrays in einer gescannten Bildebene verbessert werden.
Anregungseigenschaften von Fluorophoren
Fluorophore für Multiphotonen-Experimente sollten der gleichen Prüfung unterzogen werden wie Fluorophore für Ein-Photonen-Untersuchungen. Die Sonden sollten große Absorptionsquerschnitte bei geeigneten Wellenlängen, hohe Quantenausbeuten, eine niedrige Photobleaching-Rate und eine möglichst geringe chemische und photochemische Toxizität aufweisen. Die Fluorophore sollten zudem eine hohe Beleuchtungsintensität der Laserquelle vertragen, ohne dass es zu einer signifikanten Verschlechterung kommt. In den meisten Fällen verwenden die Forscher für Zwei-Photonen-Experimente die gleichen Fluorophore, die auch in der Weitfeld- und konfokalen Fluoreszenzmikroskopie als Marker eingesetzt werden.
Das Anregungsspektrum gängiger Fluorophore richtet sich nach dem Anregungsmodus und der Wellenlänge der einfallenden Photonen. Aufgrund dieser Abhängigkeit unterscheidet sich das Zwei-Photonen-Absorptionsspektrum (oft) deutlich von dem entsprechenden Ein-Photonen-Spektrum. Die meisten der untersuchten Fluorophore sind in der Lage, Zwei-Photonen-Anregungen bei der doppelten Wellenlänge ihres Ein-Photonen-Absorptionsmaximums zu absorbieren. Trotzdem gibt es keine fundamentale Grundlage für die quantitative Vorhersage des Zwei-Photonen-Anregungsspektrums eines komplexen Fluorophors durch die bloße Untersuchung des Ein-Photonen-Querschnitts. Zwischen den Ein- und Zwei-Photonen-Anregungsspektren hochkonjugierter unsymmetrischer Moleküle bestehen oft erhebliche Unterschiede, die in der Molekülspektroskopie genutzt werden, um Informationen über die Struktur angeregter Zustände zu erhalten. Ein gutes Beispiel sind die aromatischen Aminosäurederivate Tyrosin und Phenylalanin, deren komplexe Zwei-Photonen-Wirkungsquerschnitte sich deutlich von denen der Ein-Photonen-Anregung unterscheiden. Im Gegensatz dazu ist das Zwei-Photonen-Spektrum für Tryptophan (Abbildung 2) dem Profil für die Ein-Photonen-Anregung sehr ähnlich.
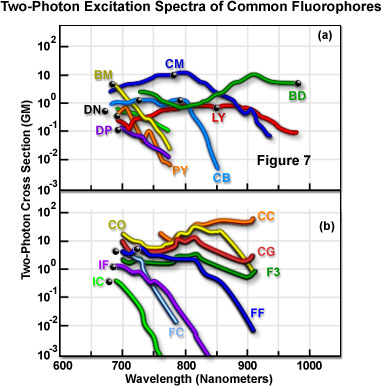
Für quantitative dreidimensionale Rekonstruktions- und Dekonvolutionsexperimente sollten die Zwei-Photonen-Absorptionsspektren von Fluorophoren gemessen werden, um sicherzustellen, dass die Anregungswellenlängen nahe der Absorptionsbänderspitzen zentriert sind. Obwohl Zwei-Photonen-Querschnitte berechnet werden können, ist der Prozess vorsichtig ausgedrückt komplex. Die direkte experimentelle Messung der Absorptionsspektren ist die bevorzugte Methode, doch sind diese Experimente aufgrund der geringen einfallenden und absorbierten Leistung und der Intensitätsschwankungen der Lichtquelle schwierig. Die Absorptionsquerschnitte wurden mit thermischen Linsen und akustooptischen Techniken bestimmt, eine möglicherweise einfachere Methode ist die Untersuchung der Photonenemission von Fluorophoren mit bekannter Quantenausbeute. Bei der Konzeption neuer Zwei-Photonen-Experimente sollte eine Reihe von Fluorophoren untersucht werden, deren Absorptionsspitzen in etwa bei der Hälfte der vorgesehenen Anregungswellenlänge liegen.
Abbildung 7 zeigt die Kennlinien der gemessenen Zwei-Photonen-Fluoreszenz-Anregungsspektren für eine Reihe von gängigen Fluorophoren. Die Daten in Abbildung 7 stellen Zwei-Photonen-Wirkungsquerschnitte dar, die sich aus dem Produkt der Fluoreszenzemission-Quanteneffizienz und dem Zwei-Photonen-Absorptionsquerschnitt ergeben. Die Spektren wurden mit linear polarisiertem Licht aufgenommen, das von einem modengekoppelten Titan-Saphir-Laser emittiert wurde. In jedem Spektrum steht der schwarze Punkt für die doppelte Wellenlänge des Ein-Photonen-Fluorophor-Absorptionsmaximums. Tabelle 1 enthält den Schlüssel der aus zwei Buchstaben bestehenden Namenscodes, die neben jedem Spektrum in Abbildung 7 angegeben sind. Die Kurven stellen die spektralen Wirkungsquerschnitte der Zwei-Photonen-Anregung der Fluorophore dar.
Zwei-Photonen-Fluoreszenz-Anregungsspektren von Fluorophoren
| Name des Fluorophors (Abkürzung) | Anregungswellenlänge (Nanometer) |
|---|---|
| (BM) p-Bis(o-methylstyryl)benzol | 691 |
| (CB) Kaskadenblauhydrazid-Trinatriumsalz | 750 |
| (YL) Luzifer-Gelb-CH-Ammoniumsalz | 860 |
| (BD - Bodipy) 4,4-Difluor-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diazaindacen-2,6-disulfonsäure-Dinatriumsalz | 920 |
| (DP - DAPI nicht DNA-gebunden) 4',6-Diamidino-2-phenylindoldihydrochlorid | 700 |
| (DN - Dansyl) 5-Dimethylaminonaphthalin-1-sulfonylhydrazin | 700 |
| (PY) 1,2-Bis-(1-pyrenedecanoyl)-sn-glycero-3-phosphocholin | 700 |
| (CM) Kumarin 307 | 776 |
| (IC) Indo-1 mit Ca++ | 700 |
| (IF) Indo-1 ohne Ca++ | 700 |
| (FC) Fura-2 mit Ca++ | 700 |
| (FF) Fura-2 ohne Ca++ | 720 |
| (CG) Calcium Green-1 mit Ca++ | 725 |
| (CO) Calcium Orange mit Ca++ | 800 |
| (CC) Calcium Crimson mit Ca++ | 850 |
| (F3) Fluo-3 mit Ca++ | 800 |
Tabelle 1
Querschnittsmessungen zeigen, dass der Anregungspeak für die Zwei-Photonen-Absorption häufig dem Profil für die Ein-Photonen-Absorption sehr ähnlich oder in Richtung blau verschoben ist (Abbildung 7). Die kürzeren durchschnittlichen Wellenlängen können bei der Kopplung der Fluorophoranregung an den verfügbaren Wellenlängenbereich der modengekoppelten Laser von Vorteil sein. Ein weiterer konsistenter Aspekt der Zwei-Photonen-Absorptionsspektren ist, dass sie in der Regel viel breiter sind als ihre Ein-Photonen-Absorptionsspektren. Dies ist vorteilhaft bei experimentellen Einschränkungen, da der Bereich der für die Anregung geeigneten Wellenlängen vergrößert wird und zwei Fluorophore, deren Zwei-Photonen-Wirkungsquerschnitte sich überschneiden, deren Ein-Photonen-Absorptionsspektren jedoch weit auseinander liegen, leichter gleichzeitig angeregt werden können. Messungen der Drei-Photonen-Wirkungsquerschnitte zeigen, dass sie im Allgemeinen den entsprechenden Ein-Photonen-Spektren sehr ähnlich sind.
Obwohl sich die Absorptionsspektren bei Ein- und Zwei-Photonen-Anregung oft unterscheiden, scheinen andere Fluoreszenzeigenschaften wie Lebensdauer, Emissionswellenlängen und Übergangsgeschwindigkeit zwischen den Systemen nicht beeinflusst zu werden. Diese Ähnlichkeit deutet darauf hin, dass sowohl durch lineare als auch durch nichtlineare Absorption dieselben Fluoreszenzanregungen erreicht werden und dass sich der Fluorophor nach der Anregung unabhängig von der Anregungsart gleich verhält. Dies gilt auch für die Drei-Photonen-Anregung, so dass die Forscher bei den meisten Mehrphotonen-Experimenten bewährte ratiometrische und spektroskopische Methoden anwenden können.
Photo- und Wärmeschäden bei Multiphotonen-Anregung
Bei allen Formen der Fluoreszenzmikroskopie werden lebende Zellen durch Licht geschädigt, wobei der Grad der Schädigung von der Anregungswellenlänge, der Expositionsdauer und den chemischen Eigenschaften der als Zellsonden verwendeten Fluorophore abhängt. Die durch die Anregungsbeleuchtung verursachten Schäden können in zwei Kategorien unterteilt werden: Wärmeschäden und Schäden durch chemische Reaktionen. Photochemische Nebenwirkungen durch biochemische Reaktionen infolge der Fluorophoranregung sind nicht gut erforscht und variieren stark zwischen verschiedenen Zell- und Gewebetypen. Wärmeschäden hingegen entstehen hauptsächlich durch zwei Mechanismen, die aufgrund der Ein-Photonen-Absorption von Wasser und die Zwei-Photonen-Absorption von Fluorophoren im Brennpunkt auftreten.
In den meisten untersuchten Zellen (insbesondere in Säugetierzellen) gibt es so gut wie keine Absorption langwelliger Nahinfrarot-Anregungsstrahlung durch intrinsische Fluorophore, wie sie in der Multiphotonen-Fluoreszenz verwendet werden. Intrazelluläres und interzelluläres Wasser, das Zellen und Gewebe umgibt, kann jedoch erhebliche Mengen an Infrarot- und Nahinfrarotlicht absorbieren und überschüssige Wärme erzeugen, die die Lebensfähigkeit biologischer Proben beeinträchtigen kann. Wird die wässrige biologische Umgebung hingegen mit den kürzeren Wellenlängen des sichtbaren und ultravioletten Spektrums beleuchtet, wie sie in der konfokalen und Weitwinkel-Fluoreszenzmikroskopie verwendet werden, wird eine erhebliche Wärmemenge nicht vom umgebenden Wasser absorbiert.
Die Erwärmung aufgrund der Ein-Photonen-Absorption durch Wasser erfolgt im gesamten Strahlengang, sowohl oberhalb als auch unterhalb der Fokusebene. Unter kontrollierten durchschnittlichen Multiphotonen-Bedingungen wurde für 700 bzw. 1000 Nanometer ein Temperaturanstieg zwischen 0,065 und 1,1 Grad Celsius berechnet. Diese Berechnungen stimmen mit Wärmemessungen überein, die mit optischer Laserpinzettenanregung bei 1064 Nanometern durchgeführt wurden. In Situationen, in denen der anregende Lichtstrahl stationär gehalten wird, kann es zu einer stärkeren Erwärmung kommen, die in einem logarithmischen Verhältnis zur Zeit schnell ansteigt. Die Erwärmung durch die Absorption des Fluorophors ist bei Experimenten mit Multiphotonen-Anregung weitgehend auf den Brennpunkt beschränkt. Die anschließende Wärmefreisetzung erfolgt gleichmäßig in einem sphärisch symmetrischen Bereich um das Fokusvolumen herum und erzeugt selbst bei hohen Fluorophor-Konzentrationen keine nennenswerte Wärmemenge.
Fazit
Die Multiphotonen-Fluoreszenzmikroskopie wird immer mehr zu einer bevorzugten Methode für die dynamische Bildgebung von Zellen und Geweben lebender Versuchstiere. Diese Technik ist besonders nützlich, wenn die Dynamik von Zellen beobachtet werden soll, die sich tief in der Probe befinden. Darüber hinaus werden bei der Multiphotonen-Anregung Nebenwirkungen wie Photobleaching und Lichtschäden minimiert und treten nur in der unmittelbaren Umgebung des Fokusvolumens auf.
Phototoxizität in Zellen ist ein kaum verstandenes Phänomen, das jedoch bei den meisten Formen der Fluoreszenzmikroskopie verstärkt auftritt. Die niedrigere Quantenenergie und die geringere intrinsische Absorption längerer Wellenlängen in der Multiphotonenmikroskopie verringern die schädlichen Auswirkungen des Lichts auf lebende Zellen und Gewebe und ermöglichen so die Untersuchung der Zelldynamik. Ein Haupthindernis für die Forschung im Bereich der Multiphotonenmikroskopie sind die hohen Kosten für die Technik, insbesondere für die modengekoppelten gepulsten Lasersysteme, die für die Zwei- und Drei-Photonen-Anregung benötigt werden. Die beliebtesten ultraschnellen Lasersysteme sind Titan-Saphir-Laser. Durch die Abstimmbarkeit der Wellenlänge ist der gepulste Titan-Saphir-Laser (700 bis 1300 Nanometer) vielseitiger einsetzbar. Diese Verfügbarkeit dürfte die breite Anwendung dieser Technik in den Biowissenschaften fördern.
Sorry, this page is not
available in your country.



