










Olympus Microscopy
Explore our wide range of systems for all your application needs


Featured Pages
Evident Image of the Year Award
Evident has launched its fifth Global Image of the Year Scientific Light Microscopy Award, recognizing the best in life science imaging worldwide.

New Investigator Program
Setting up a new lab? Contact us to find out how Olympus' New Investigator program can help!
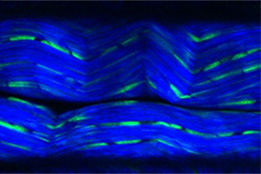
Overcome the Boundaries of 3D Live Cell Imaging
3D cell culture models are rapidly gaining traction in drug discovery studies. Find out how Olympus and scientists from Novartis developed a breakthrough method for imaging these models in multiwell plates using multiphoton microscopy.

The Evident Discovery Center
The Evident Discovery Center program reflects one of the core philosophies of Evident – that of supporting researchers by providing innovative microscopy equipment to help them push forward the frontiers of science.

Objective Finder
Select the right objective for your application from Olympus' broad line of objectives.
.jpg?rev=4DE5)
Evident Techno Lab
Evident Techno Lab is a space where you can experience the model you are considering purchasing and learn the correct way to use the microscope through the microscope classroom. (Advance reservation is required)

Microscopy Resource Center
The microscope resource center offers a variety of content at all levels, from basic microscopy concepts to specialized techniques.

Let the World See Your Work
Do you enjoy capturing images with your Olympus microscope? We’re always looking for beautiful and interesting work to share on our Olympus Life Science social media pages.