Specimen Preparation Using Synthetic Fluorophores and Immunofluorescence
Indirect Single, Double, and Triple Immunofluorescence with Brain Tissue Floating Cryosections
Indirect (two-stage) immunofluorescence is a powerful imaging technique that enables the visualization of highly specific targets using a combination of primary (direct) and secondary (indirect) antibodies. Antigenic determinants of interest in brain tissue are first treated with a monoclonal or polyclonal primary antibody from a common host (mouse, rabbit, chicken, rat, donkey, etc.) followed by a secondary antibody directed to the host primary and conjugated to a synthetic or natural fluorophore. Among the antibody targets often labeled in brain tissue sections are glial fibrillary acidic protein (GFAP), neuronal and axonal neurofilaments (both phosphorylated and non-phosphorylated; heavy, medium, and light), class III beta-tubulin, microtubule-associated proteins, blood-brain barrier proteins, synaptic proteins, myelin, and a host of antigens associated with specific diseases.
Among the useful fluorescent markers for visualization of antigens in brain tissues are rhodamine, fluorescein, the Alexa Fluor series, and the cyanine dyes. Counterstaining for nuclei using a variety of popular DNA-binding dyes follows treatment with the secondary antibody. This protocol details a generalized procedure for staining frozen brain floating cryosections between 30 and 50 micrometers in thickness.
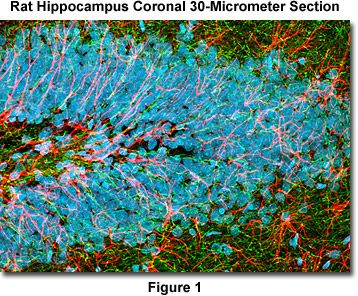
Presented in Figure 1 is a confocal image revealing the intricate network structure of glial fibrillary acidic protein and heavy neurofilaments in a thick (30-micrometer) section of rat brain hippocampus (coronal section). The section was stained as detailed below using a cocktail of chicken anti-NF-H and rabbit anti-GFAP primary antibodies, followed by goat anti-chicken and anti-rabbit secondary antibodies conjugated to the synthetic fluorophores Alexa Fluor 488 and Alexa Fluor 568, respectively. Nuclei were counterstained with DRAQ5. The specimen was imaged with a 60x oil immersion objective (without zoom) using a 488-nanometer argon-ion laser (Alexa Fluor 488), a 543-nanometer helium-neon laser (Alexa Fluor 568), and a 633-nanometer helium-neon laser (DRAQ5; pseudocolored cyan). The brain section image was acquired in grayscale and subsequently pseudocolored with hues approximating the fluorescence emission spectra of the respective probes, with the exception as noted above.
Reagents
- Dulbecco's Phosphate Buffered Saline (PBSA) - Dissolve 0.2 grams of potassium chloride, 0.2 grams of monobasic potassium phosphate, 8.0 grams of sodium chloride, and 1.74 grams of dibasic sodium phosphate (heptahydrate) in one liter of double-distilled water. Adjust the pH to 7.2 with concentrated sodium hydroxide.
- Phosphate Buffered Saline with Calcium and Magnesium (PBS) - Dissolve 0.2 grams of potassium chloride, 0.2 grams of monobasic potassium phosphate, 8.0 grams of sodium chloride, and 1.74 grams of dibasic sodium phosphate (heptahydrate) in one liter of double-distilled water. After dissolving these reagents add 0.132 grams of calcium chloride dihydrate and 0.10 grams of magnesium chloride hexahydrate. Adjust the pH to 7.2 with concentrated sodium hydroxide. Addition of the divalent alkaline earth metals to the buffer solution is helpful to ensure that uncondensed chromatin remains intact and contained within the nucleus during the staining procedure, dramatically decreasing background fluorescence levels when using DNA probes excited by ultraviolet irradiation (DAPI and Hoechst).
- Fixative - Dissolve 3.7 grams of paraformaldehyde in 100 milliliters of PBSA with mild heating (prepare fresh fixer each day). After cooling, filter the solution.
- Permeabilization Buffer - 0.2 percent Triton X-100 in PBS (sonicate detergent buffer for 30 minutes to one hour immediately before use).
- Blocking Buffer - 10 percent normal goat serum (NGS) in PBS containing 0.05 percent Triton X-100 (add 2-3 milligrams sodium azide per 100 milliliters of blocking buffer to eliminate the growth of microorganisms). If secondary antibodies to a host other than goat are being used, prepare the Blocking Buffer with normal serum from that species.
- PBS Wash Buffer - Use PBS buffer after fixation, before permeabilization and after treatment with the primary and secondary antibody probes immediately prior to nuclear staining except for those tissues requiring specialized buffers for nuclear dyes that are not compatible with PBS.
- PBS-Triton Wash Buffer - For wash sequences immediately after permeabilization and after antibody treatment (before staining with nuclear dyes), use PBS containing 0.05 percent Triton X-100.
- PBS-Triton Wash Buffer with Blocking Serum - For wash sequences between the primary and secondary antibody incubations and immediately after the secondary antibody treatment, use PBS with 0.05 percent Triton X-100 and 1 percent normal host (goat) serum.
- Primary Antibody Cocktail - Add the appropriate volume of concentrated primary antibody stock solution to Blocking Buffer diluted 50-percent with PBS-Triton wash buffer (to yield a final concentration 5-percent normal goat serum). Several primary antibodies from different hosts can be mixed into a cocktail. Tissue thin sections receive 250-500 microliters of the antibody solution.
- Secondary Antibody/Phalloidin Cocktail - Add the appropriate volume of secondary antibody conjugated to the selected fluorophore (for example, 8 microliters of goat anti-mouse heavy and light chain secondary IgG with Alexa Fluor 350 at 2 milligrams per milliliter) and 20-30 microliters of phalloidin stock solution (6.6 micromolar) to 1 milliliter of Blocking Buffer diluted 50-percent with PBS-Triton wash buffer (to yield a final concentration 5-percent normal goat or other host serum). In many cases, the phallotoxin actin probe can be substituted with one or more additional secondary antibodies from other species (matched to the primary antibodies). As with the primary antibody mixture, tissue thin sections receive 250-500 microliters of the secondary antibody solution.
- Nuclear Dye - Prepare fresh dilutions of the nuclear dye immediately prior to staining. When using SYTOX, DRAQ5, and the cyanine nuclear stains, background fluorescence can be significantly reduced (especially in the cytoplasm) by adding 10 milligrams of RNase to the Blocking Buffer. In this case, block the cells or tissues at 37 degrees Celsius, rather than at room temperature, to activate the enzyme.
- Nuclear Dye Wash Buffer - Hoechst and SYTOX dyes require Hanks Balanced Salt Solution (Hanks BSS), while DAPI, as well as the monomeric and dimeric cyanine nuclear stains, can be used with PBS.
Nuclear Counterstain Dilutions
- Hoechst (33342 and 33258) - Dilute 5 microliters of 10 milligram/milliliter stock solution in 150 milliliters of Hanks BSS (treat for 30 minutes).
- SYTOX Green and Orange - Dilute 10 microliters of concentrated stock solution (5 millimolar in dimethyl sulfoxide) in 250 milliliters of Hanks BSS (treat for 30 minutes).
- DAPI - Dilute 5 microliters of 10 milligram/milliliter stock solution in 150 milliliters of PBS diluted 50-percent with double-distilled water (treat for 5 minutes).
- Monomeric and Dimeric Cyanine Dyes - Dilute the concentrated stock solution (for example, TO-PRO-3; usually 1 millimolar) as recommended by the manufacturer (1:20 to 1:100) into PBS (treat for 5 to 30 minutes).
- DRAQ5 - Dilute the concentrated stock solution (usually 1 millimolar) as recommended by the manufacturer (1:20 to 1:100) into PBS (treat for 5 to 30 minutes).
Procedure
Transfer one or more frozen (-80 degrees Celsius) floating cryosections to a 6-well culture dish at freezer temperature and place on dry ice for transport. Gently add 2 milliliters of PBS buffer to each well containing a section to allow the sections to thaw and float in the buffer. Slowly bring the culture dish to room temperature and rotate the tissue sections as they are being hydrated on an orbital shaker at 5-10 revolutions per minute.
Note: Do not allow sections to warm to room temperature before adding the PBS buffer. Carefully aspirate all solutions described below away from the floating sections with a Pasteur pipette and a squeeze bulb (do not use a vacuum aspirator).
Floating sections were fixed before removal from the animal but can be fixed again after rehydration using paraformaldehyde treatment for 30 minutes. This is an optional step.
Wash each section 4 times in PBS for 15 minutes (each wash). Slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Dissolve the membranes in the tissue sections with Permeabilization Buffer by treatment for two hours. Slowly rotate the tissue sections as they are being permeabilized on an orbital shaker at 5-10 revolutions per minute.
Block nonspecific secondary antibody and synthetic fluorophore binding sites with Blocking Buffer for 2-4 hours at 37 degrees Celsius.
After blocking, carefully add the primary antibody cocktail (1 milliliter per culture chamber well) without disturbing the tissue section. Incubate the covered culture plate in a polyethylene bag in the refrigerator (4 degrees Celsius) for 48 hours. It is not necessary to cover the culture chambers with aluminum foil (to protect from light) at this point because fluorophores are not present.

On the second day after addition of the primary antibodies, wash the floating sections four times (20 minutes each wash) with PBS-Triton Wash Buffer containing 1 percent Blocking Serum. Slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
After the wash sequence, carefully add the secondary antibody cocktail (1 milliliter per culture chamber well) without disturbing the tissue section. Incubate the covered culture plate in a polyethylene bag in the refrigerator (4 degrees Celsius) for 48 hours. Protect from light by placing a sheet of aluminum foil over the culture chamber.
On the second day after treatment with the secondary antibodies, wash the floating sections four times (20-minutes each wash) with PBS-Triton Wash Buffer containing 1 percent Blocking Serum. Cover the culture chamber with aluminum foil (to protect from light) and slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Completely remove blocking serum wash buffer by washing the tissue sections with PBS for two or three buffer exchanges (15-minutes each wash).
For DAPI and cyanine nuclear counterstains, add the diluted dye in PBS to the culture chamber well and treat the specimen for the recommended time: 5-10 minutes for DAPI; 15-30 minutes for cyanine dyes (protect from light with aluminum foil). When using Hoechst or SYTOX stains (30-minute incubation), first wash the slides in Hanks Balanced Salt Solution for two buffer exchanges prior to counterstaining.
Wash the counterstained specimens with either PBS or Hanks Balanced Salt Solution (depending upon the nuclear dye) for three times at 15 minutes for each wash. Protect from light with aluminum foil.
After the final washing step, place a 22 × 22 millimeter (or 22 × 50 millimeter, depending upon the section size) coverslip under each section and carefully aspirate the buffer with a pipette while simultaneously positioning the glass under the section so that it adheres to the center of the coverslip. After the buffer has been completely removed, pull the coverslip out from the well with the aid of forceps. Place the coverslip on a flat surface, add one drop of mounting media, place a cleaned microscope slide on the section and turn the slide with the coverslip up-side down. Leave slides at room temperature for at least 1-2 hours to dry the sections.
Sorry, this page is not
available in your country.



