When specimens, living or non-living, organic or inorganic, absorb and subsequently re-radiate light, the process is described as photoluminescence. If the emission of light persists for up to a few seconds after the excitation energy (light) is discontinued, the phenomenon is known as phosphorescence. Fluorescence, on the other hand, describes light emission that continues only during the absorption of the excitation light. The time interval between absorption of excitation light and emission of re-radiated light in fluorescence is of extraordinarily short duration, usually less than a millionth of a second.
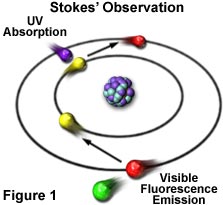
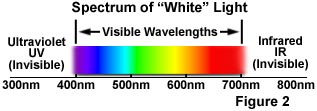
The phenomenon of fluorescence was known by the middle of the nineteenth century. British scientist Sir George G. Stokes first made the observation that the mineral fluorspar exhibits fluorescence when illuminated with ultraviolet light, and he coined the word "fluorescence". Stokes observed that the fluorescing light has longer wavelengths than the excitation light, a phenomenon that has become to be known as the Stokes shift. In Figure 1, a photon of ultraviolet radiation (purple) collides with an electron in a simple atom, exciting and elevating the electron to a higher energy level. Subsequently, the excited electron relaxes to a lower level and emits light in the form of a lower-energy photon (red) in the visible light region. Figure 2 is a diagrammatic representation of the visible light region of electromagnetic radiation, which covers a wavelength range from approximately 400 to 700 nanometers. Surrounding the visible region is higher energy ultraviolet light and lower energy infrared light.
Fluorescence microscopy is an excellent method of studying material that can be made to fluoresce, either in its natural form (termed primary or autofluorescence) or when treated with chemicals capable of fluorescing (known as secondary fluorescence). The fluorescence microscope was devised in the early part of the twentieth century by August Köhler, Carl Reichert, and Heinrich Lehmann, among others. However, the potential of this instrument was not realized for several decades, and fluorescence microscopy is now an important (and perhaps indispensable) tool in cellular biology.
Early investigations revealed that many specimens, including microminerals, crystals, resins, crude drugs, butter, chlorophyll, vitamins, and inorganic compounds, exhibit autofluorescence when irradiated with ultraviolet light. However, it was not until the 1930's that Austrian investigator Max Haitinger and other scientists developed the technique of secondary fluorescence, which employs fluorochrome stains to label specific tissue components, bacteria, and other pathogens that do not autofluoresce. These fluorochrome stains, tagged to specific biochemical targets, spurred the use of the fluorescence microscope. The instrument's value was significantly enhanced by the 1950's when Albert Coons and Nathan Kaplan demonstrated the localization of antigens in tissues that were reacted with a fluorescein-labeled antibody.
Electron Excitation and Emission
Discover how electrons are elevated to higher energy states from which they fluoresce when their associated atoms are irradiated by photons of varying energy levels.
The basic task of the fluorescence microscope is to permit excitation light to irradiate the specimen and then to separate the much weaker emitted fluorescent light from the brighter excitation light. Thus, only the emission light from the specimen reaches the eye or other detector (usually a digital or conventional film camera). The resulting fluorescing areas shine brightly against a dark background with sufficient contrast to permit detection. The darker the background behind the non-fluorescing material, the more efficient the instrument.
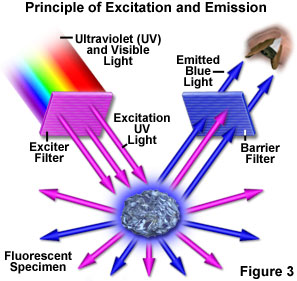
Figure 3 presents a graphical representation of the occurrences when a fluorescent specimen is observed with a fluorescence microscope. Ultraviolet (UV) light of a specific wavelength or set of wavelengths is produced by passing light from an ultraviolet-emitting source through the exciter filter. The filtered ultraviolet light illuminates the specimen, in this case a crystal of fluorspar, which in turn emits fluorescent light when illuminated by the ultraviolet light. Visible light emitted from the specimen, red in Figure 3, is then filtered through a barrier filter that does not allow reflected ultraviolet light to pass. It should be noted that this is the only mode of microscopy in which the specimen, subsequent to excitation, produces its own light. The emitted light re-radiates spherically in all directions (over a 360-degree angle), regardless of the direction of the exciting light.
Fluorescence microscopy is a rapidly expanding and invaluable tool of investigation. Its advantages are based upon attributes not as readily available in other optical microscopy techniques. The use of fluorochromes has made it possible to identify cells and sub-microscopic cellular components and other entities with a high degree of specificity amidst non-fluorescing material. What is more, the fluorescence microscope can reveal the presence of fluorescing material with exquisite sensitivity. An extremely small number of fluorescent molecules (as few as 50 molecules per cubic micrometer) can be detected. In a given sample, through the use of multiple staining, different probes will reveal the presence of individual target molecules. Although the fluorescence microscope cannot provide spatial resolution below the diffraction limit of the respective specimens, the presence of fluorescing molecules below such limits is made remarkably visible.
The techniques of fluorescence microscopy can be applied to organic material, formerly living (biological) material, or to living material (with the use of in vitro or in vivo fluorochromes) or to inorganic material (especially in the investigation of contaminants on semiconductor wafers). There are also a burgeoning number of studies using fluorescent probes to monitor rapidly changing physiological ion concentrations, such as calcium and magnesium, and pH values in living cells.
Many plant and animal tissues, as well as materials specimens, inherently fluoresce when irradiated with shorter wavelength light (primary or autofluorescence). Autofluorescence has been found useful in plant studies, coal petrography, sedimentary rock petrology, and in the semiconductor industry. In the study of animal tissues or pathogens, autofluorescence is often either extremely faint or of such non-specificity as to make autofluorescence of minimal utility. Of far greater value for such specimens are the extrinsic fluorochromes (also called fluorophores) that are excited by irradiating light, and whose eventual yield of emitted light is of greater intensity. Such fluorescence is known as secondary fluorescence.
Fluorochromes are stains, somewhat similar to the better-known visible light absorbing tissue stains, which attach themselves to visible or sub-visible organic matter. These fluorochromes, capable of absorbing and then re-radiating light, are often highly specific in their attachment targeting and have significant yield in absorption-emission ratios (a concept termed quantum yield). This makes fluorochromes extremely valuable in biological applications. The growth in the use of fluorescent microscopes is closely linked to the development of hundreds of fluorochromes with known intensity curves of excitation (absorption) and emission and well-understood biological structure targets.
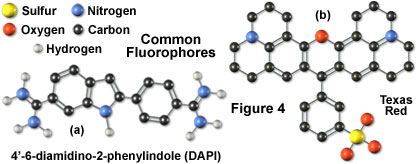
Illustrated in Figure 4 are two of the most commonly used fluorochromes in fluorescence microscopy. The popular nucleic acid stain 4',6-diamidino-2-phenylindole (DAPI), the left-hand molecule in Figure 4, is an indole derivative featuring two highly nucleophilic amidino moieties, which is useful for fluorescent labeling of nucleic acids and has been widely used in the fluorescence analysis for rapid identification of pathogens. Originally synthesized as a potential anti-trypanosomal agent, DAPI binds preferentially to adenosine and thymidine (A-T) base pair regions in DNA and is excited by ultraviolet light with a maximum absorption wavelength of 355 nanometers. The dye fluoresces in the blue region of the visible light spectrum. To the right of the DAPI molecule (Figure 4) is another fluorochrome, Texas Red, whose properties in fluorescent conjugates have been thoroughly studied. This fluorochrome was developed for double labeling in an application known as flow cytometry, but is extensively used in place of rhodamine (another common fluorochrome) for antibody detection in fluorescence microscopy.
In many cases, Texas Red is employed in combination with DAPI and fluorescein isothiocyanate (FITC) for multiply stained specimens that can be observed due to the red, blue, and green fluorescence of the dyes. When deciding which label to use for fluorescence microscopy, it must be borne in mind that the chosen fluorochrome should have a high likelihood of absorbing the exciting light and should remain attached to the target molecules. The fluorochrome should also be capable of providing a satisfactory yield of emitted fluorescence light.
One of the most important applications of fluorescence microscopy is in the field of immunofluorescence. A living animal manufactures innumerable antibodies that are used, in conjunction with white blood cells, to neutralize any entering foreign bodies, such as viruses, bacteria, and foreign proteins, which contain or produce antigens. The antigen-antibody reaction is highly specific, often likened diagrammatically to a lock and key relationship. Immunofluorescence owes its success to the marriage between the sensitivity of fluorescence microscopy and the high degree of specificity exhibited by immunology.
In direct immunofluorescence, a specific antibody is labeled by chemically attaching a fluorochrome to form what is known as a conjugate, which is then spread on a microscope slide containing the suspected presence of a particular antigen known to stimulate production of the antibody. If the antigen is present, the labeled antibody conjugate binds to the antigen and remains bound to the specimen after it is washed. The presence of the chemically attached fluorescent conjugate and antigen is demonstrated when the fluorochrome is excited at its excitation peak, and the subsequent emission intensities at various wavelengths can then be observed visually or captured by a detector system (digital or traditional camera).
Another commonly used technique is known as indirect immunofluorescence, where serum possibly containing unlabeled antibody and its related, but known, antigen are incubated together. A fluorochrome conjugated to an anti-human antibody (if the specimen being tested is human) is then placed on the slide containing the unlabeled antibody-antigen. If indeed, there has been an antigen-antibody reaction, the fluorochrome-labeled anti-human antibody attaches itself to the complex formed by the antigen and antibody. Subsequently, the labeled grouping of antigen, antibody, and fluorochrome labeled anti-human antibody is excited at the peak wavelength intensity for that fluorochrome and any resulting emission is observed. The indirect immunofluorescence technique reduces the necessity of keeping in stock large numbers of labeled antibodies, and also usually results in greater fluorescence intensity.
A significant area of fluorescence investigation deals with cytochemical and histochemical staining. Fluorochromes have been used to identify chromosomes, DNA content, proteins, cellular structures, hormones, and vitamins. The fluorescence microscope is a powerful tool in such studies because of the exquisite sensitivity of selected fluorochromes and their specificity for extremely minute quantities in the specimen. Indeed, although the fluorescence microscope is limited in its spatial resolution to the usual rules governed by numerical aperture and diffraction limits, fluorescence probes can, through emitted light, reveal the presence of fluorescing material by making such material visible even when present in only sub-resolution quantities (such as a few molecules).
A burgeoning group of applications for fluorescence microscopy involve the application of fluorescent probes (fluorochromes) to living materials, both in vitro (in glass) and in vivo (in life). The difficulties multiply for these probes because of constraints imposed by possible toxicity. In addition, a considerable amount of attention must be paid to time intervals because of the ever-changing nature of the life processes and movement of intracellular structures. Fluorescence investigations have been applied to changes in ion concentration, bound and unbound, for important biological ions such as hydrogen (pH), calcium, and magnesium.
Among the best known are studies of intracellular calcium using the popular fluorescent probe Fura-2. For this dye, dual excitation at approximately 340 and 380 nanometers (using a light chopper and dual excitation filters or monochromators) can be monitored at a single emission wavelength, which is measured independently for each excitation band. A host computer controlling the microscope is used to calculate the ratio (in a process known as ratio imaging fluorescence) of bound to free intracellular calcium as revealed by the changes in fluorescence emission intensity. The advantage of the ratio method is that essentially all factors are kept constant except the dual near-ultraviolet excitation wavelengths, each of which produce emission in the green part of the visible light spectrum. A similar type of ratio imaging is conducted with a probe known as Indo-1. For this fluorochrome, also used for determination of intracellular bound and unbound calcium, a single excitation wavelength is employed, but emission is measured at each of two wavelengths to distinguish bound from unbound calcium.



