TruAI™ 딥 러닝 기술을 사용한 효모 단백질 위치 정보 분류
시험대에 오른 인공지능(AI)
인공지능(AI) 기술은 대량의 현미경 이미징 데이터를 처리해야 하는 연구자들의 수작업 부담을 완화하는 데 도움을 주고 있습니다. 훈련된 TruAI™ 딥 러닝
신경망은 복잡한 데이터 세트의 자동 객체 분할을 지원하지만, 조정 가능성과 효율성이 얼마나
뛰어날까요? 이 애플리케이션 노트는 특히 어려운 응용 분야인 효모 단백질 위치 정보를 분류하는 고함량 분석에 대한 TruAI 기술의 성능을 검증합니다.
과제: 효모 분류를 위한 고함량 형광 스크리닝
단백질의 세포 내 위치 파악은 단백질의 생물학적 기능을 이해하기 위한 필수 전제 조건입니다. 모델 유기체인 사카로미세스 세레비시아(Saccharomyces cerevisiae)(이제부터 효모로 지칭)에서 단백질의 위치 정보를 살펴보기 위해 과학자들은 게놈 전반에서 포괄적인 돌연변이 모음을 얻었습니다. 이러한 돌연변이에는 N-[1,2] 또는 C-말단[3,4]에 형광 표지자로 표지할 수 있는 단백질이 있습니다. 연구자들은 이들의 형광 패턴을 연구하여 현미경으로 돌연변이 단백질의 위치를 관찰할 수 있습니다(그림 1 참조).
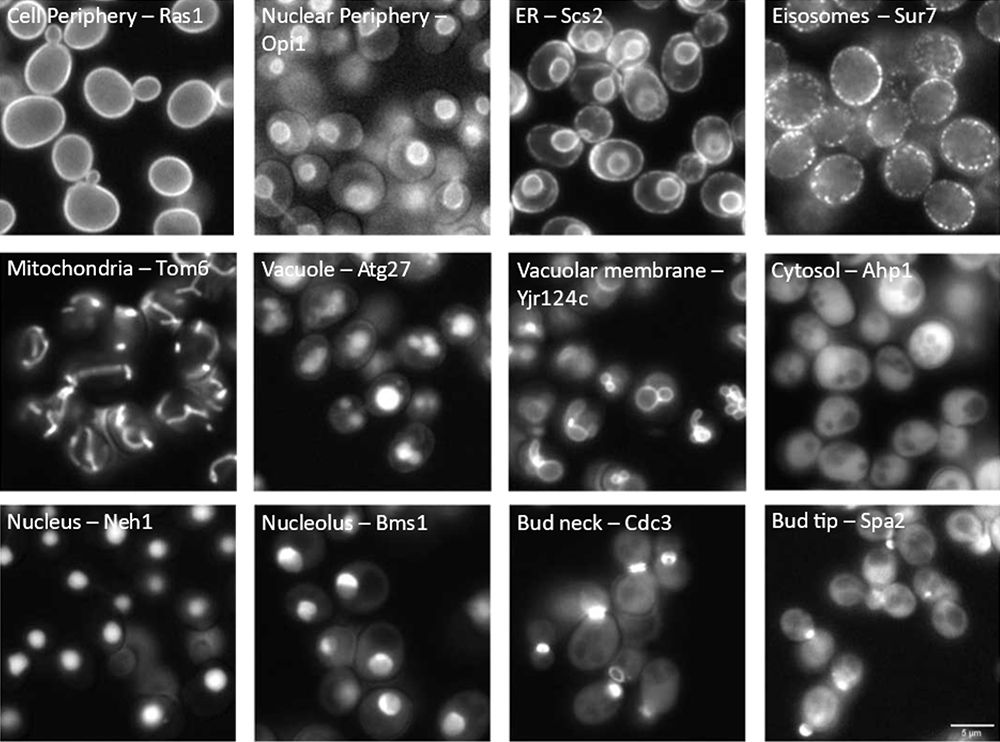
그림 1: 형광 태그가 지정된 효모 단백질의 세포 구획 위치 파악 시각화된 구획(왼쪽) 및 태그가 지정된 각 단백질의 표준 이름(오른쪽)이 각 이미지에 라벨링되어 있습니다.
효모는 대략적으로 총 6000개의 유전자를 갖고 있습니다. 따라서, 이러한 게놈 전반의 돌연변이 모음에는 약 6000가지의 개별 돌연변이종이 필요합니다. 특정 과학적 질문에 맞게 모음을 만들기 위해 수천 개의 효모종의 유전자 조작을 동시에 수행할 수 있도록 고함량 접근법이 개발되었습니다. 예를 들면, 게놈 전반의 형광 돌연변이 모음에 유전자 삭제를 도입하거나, 두 번째 형광 표지자를 도입하여 서로 다른 단백질 간 공간적 관계를 평가했습니다. 이처럼 대규모의 돌연변이 모음을 효과적으로 처리하고 자동화된 현미경 검사로 이미지를 획득하기 위한 효율화된 접근법이 있습니다[5]. 그러나 이러한 고함량 분석에서 병목 지점은 형광 패턴에 따라 단백질 위치를 파악하기 위해 생성된 대량의 이미지 데이터를 분석하는 것입니다.
이 애플리케이션 노트에서는 형광 패턴을 기반으로 AI 모델을 만들어 다양한 효모종의 다양한 세포 구획에서 단백질 위치를 자동으로 분류하기 위해 scanR
고함량 스크리닝 플랫폼과 함께 TruAI™ 딥 러닝 기술을 사용하는 방법을 보여드릴 것입니다.(그림 1).
이상값 주석 및 AI 훈련
어느 AI 모델을 개발하든, 첫 번째 단계는 이미지의 특정 픽셀 패턴을 특정 클래스와 연관시키는 이상값(ground truth)을 생성하는 것입니다. 표준 이미지 분할 작업의 경우, 소프트웨어 라벨링 도구를 사용한 수작업 주석 추가를 통해 이를 쉽게 수행할 수 있습니다[6]. 그러나 모델이 구분해야 하는 클래스가 많을수록 더 많은 이상값 주석을 추가해야 하므로 사용자의 주석 추가 부담이 증가하고 수작업 주석 추가 효율성이 감소하고 시간이 오래 걸릴 수 있습니다.
다양한 변종을 기반으로 효과적인 일반화를 수행하고 이미징 조건의 변화에 따라 조정 가능한 모델을 생성하는 것이 목표일 경우 이 작업은 훨씬 더 어려워집니다. 이러한 변화의 예로는 초점 품질, 형광 대조, 신호 대 잡음비 등이 있으며, 이 모든 변경 사항은 주석을 통해 적절하게 설명되어야 합니다.
이러한 문제를 해결하기 위해, 우리는 TruAI 도구를 원활하게 통합하는 scanR 고함량 스크리닝 소프트웨어와 함께 스마트 샘플 준비 방법을 활용했습니다. 이 소프트웨어는 이상값 주석 추가 작업의 자동 할당을 효율화하여 이 프로세스를 대폭 간소화합니다.
우리는 384 웰 플레이트를 사용하여 위치가 해당되는 것으로 알려진 형광 표지 단백질을 표현하는 다양한 돌연변이종을 준비하여 이미지화했습니다. 그리고 총 12가지의 위치(세포 주변부, 핵 주변부, 소포체(endoplasmic reticulum, ER), 아이소솜(eisosomes), 미토콘드리아, 액포, 액포막, 시토솔, 핵, 핵소체, 버드 넥 및 버드 팁)를 위한 몇 가지 대표자를 선택했습니다. 각 위치 클래스 내 표현형 가변성을 얻기 위해, 각 위치에 대해 다수의 독립적 변종을 선택하여 총 133개 변종을 훈련에 사용했습니다(그림 2).

그림 2: 이상값 샘플 준비를 위한 384 웰 플레이트의 레이아웃 각 웰은 특정 단백질이 N-말단에 GFP로 태그가 지정된 변종에 해당됩니다. 동일한 행의 모든 변종은 같은 단백질 위치를 공유하며, 동일한 클래스의 이상값에 할당되었습니다.
scanR 광시야 현미경 및 40배 공기 대물렌즈(NA 0.95)를 사용하여 이미지화를 수행했습니다. 단일 효모 세포를 식별하기 위해, 소프트웨어의 내장형 사전 훈련 AI 모델을 사용하여 전달 채널에서 분할을 수행했습니다[7](그림 3). 아티팩트와 비정상 세포를 제외하기 위해, 결과적으로 얻어진 객체를 순환성 요소와 영역으로 필터링했습니다. 모든 분할 마스크는 소속 웰(즉, 클래스 및 변종) 및 정상 또는 비정상 세포로 필터링 여부 등 각 단일 분할 효모에 대한 매개변수 정보를 포함한 단일 파일에 자동으로 저장됩니다. 이 파일은 정상 세포의 12개 클래스에 대해 이상값 주석을 생성하기 위해 scanR 소프트웨어의 TruAI 인터페이스 내에서 사용됩니다. 정상 세포로 필터링된 모든 픽셀은 훈련을 위해 무시됩니다. 이러한 방식으로, 여러 가지 변종 및 다양한 일반적인 이미지 변화(초점, 이미지 대조, 신호 강도, 세포 잔해 등)를 반영하는 4000~15,000개의 개별 주석을 획득했습니다.
모든 분할을 완료하고 이상값을 할당한 후, TruAI 훈련 구성을 설정했습니다. 우리는 네트워크 일반화(Generalizing Network) 및 의미론적 분할(Semantic Segmentation) 옵션을 선택하고, 픽셀 클래스 중첩을 지원했으며, 350,000회 반복에 걸쳐 훈련을 수행했습니다.
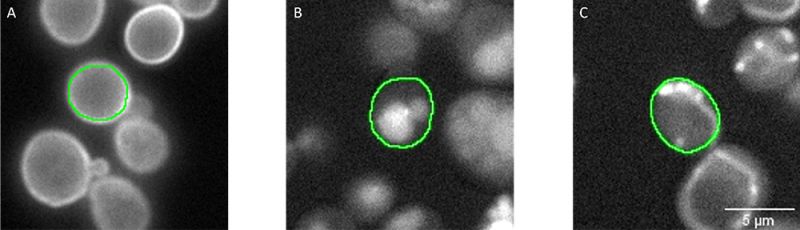
그림 3: 전달 채널(표시되지 않음)에서 분할된 개별 세포의 분할 마스크(녹색). 형광 신호는 A) 세포 주변부(웰 60, C12), B) 액포(웰 266, L2) 및 C) 미토콘드리아(웰 147, G3)에서
위치가 파악되는 단백질을 나타냅니다. 분할 마스크, 형광 채널, 웰 번호를 결합하여 scanR 소프트웨어에서 이상값 주석을 자동으로 할당했습니다.
결과 및 AI 기반 분류 솔루션 검증
실제 성능을 평가하기 위해 훈련에 포함되지 않았던 독립적 데이터 세트를 사용하여 모델을 평가했습니다. 우리는 12가지 단백질 위치 클래스에 속한 형광 태그 지정 단백질을 표현하는 변종으로 새로운 384 웰 플레이트를 준비했습니다. 전달 및 형광을 통해 이미지화한 후, 두 가지 AI 모델(전달 상태의 세포 감지를 위한 내장형 사전 훈련 AI 모델 및 형광 패턴에 따라 단백질 위치를 분류하기 위한 새 모델)을 적용하여 scanR 소프트웨어에서 자동 분석을 수행했습니다. 성능 결과를 신속하게 시각화하기 위해, 제시된 예와 같이 핵소체로 위치가 파악되는 단백질 식별을 위해 특정 클래스에 대한 확률이 높은 세포의 비율을 각 웰에 표시하는 히트 맵을 소프트웨어에서 생성하고, 이러한 단일 세포의 갤러리를 생성할 수 있습니다(그림 4).
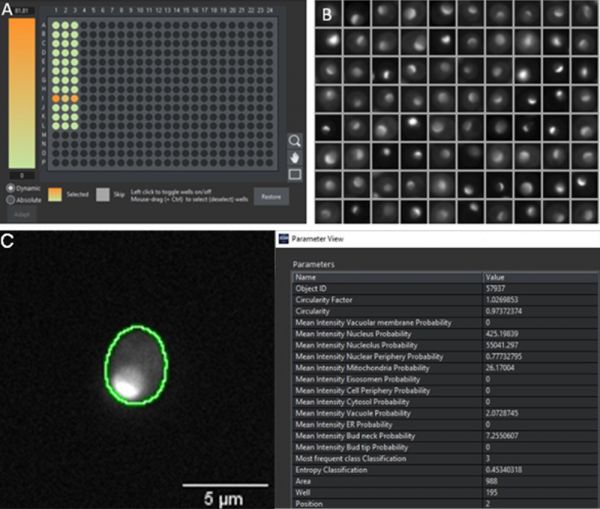
그림 4: A) 핵소체 클래스에 속한 세포의 확률을 웰에 표시하는 384 웰 플레이트 전반의 히트 맵. 히트 맵은 행 I에서 단백질이 핵소체로만 위치가 파악되는 것을 보여줍니다. B) 핵소체 위치에 해당되는 형광 패턴을 시각적으로 확인해 주는 높은 핵소체 확률을 가진 분할된 객체의 갤러리(그림 1과 비교). C) 분할 마스크에서 추출된 매개변수 세트가 있는 웰 I3의 분할된 세포. 이 예에서 핵소체는 가장 높은 값(55041)을 갖고 있습니다. 이는 두 번째로 높은 점수 클래스보다 100배 이상 높은 수치입니다(핵, 값 425).
모델 성능을 더 정밀하게 평가하기 위해, AI 분류 예측을 이상값 주석과 비교했습니다. 이는 혼동행렬에 묘사되어 있습니다(그림 5).
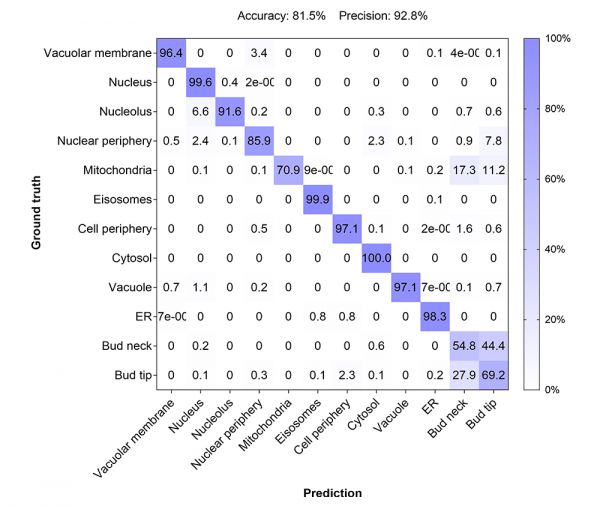
그림 5: 예측 클래스와 실제 클래스를 비교하여 개발된 AI 모델의 성능을 평가하기 위한 혼동행렬(클래스당 1600~4000개의 개별 세포).
이 행렬은 다음으로 정의되는 81.5%의 전체 정확도와 92.8%의 정밀도를 제공했습니다.
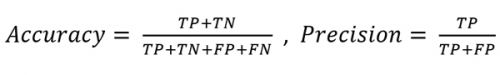 |
TP: True positive(진양성)
|
이 모델은 버드 넥 및 버드 팁 클래스를 제외하고 모든 단백질 위치에 대해 우수한 성능을 보여주었습니다. 모델은 위치가 버드 넥 또는 버드 팁이라고
예측했지만, 이 두 위치를 구분하는 데 어려움을 겪었습니다. 세포 주기 단계에 따라 두 클래스는 상당한 위치 중첩을 나타내므로 이러한 한계는
생물학적 원인 때문일 수 있습니다(그림 1 참조).
결론: 고함량 AI 스크리닝 효모 단백질 위치 파악의 장점
우리는 스마트 샘플 준비 및 수천 개의 세포로의 이상값 자동 할당을 통해 효모에서 단백질 위치의 10가지 클래스를 정확하게 예측할 수 있는 AI 딥 러닝
모델 개발이 가능함을 보여주었습니다. 이 방법론은 다른 복잡한 분류 훈련에 적용할 수 있으며, 다음과 같은 장점을 제공합니다. 1) 사용자에게
소프트웨어 프로그래밍 능력을 요구하지 않음, 2) 시간이 오래 걸리는 수작업 주석 추가를 방지하여 시간을 절약, 3) 폭넓은 이상값 주석 작업 덕분에
이미징 조건 변화에 맞는 AI 분류 네트워크를 만들 수 있는 잠재력이 있어 고함량 스크리닝 응용 분야를 포함하여 많은 샘플에 일괄적으로 적용하기에
적합함.
참고 문헌
- Yofe, I. et al. (2016) One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat. Methods 13, 371–378
- Weill, U. et al. (2018) Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods 15, 617–622
- Huh, W.-K. et al. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–91
- Meurer, M. et al. (2018) Genome-wide C-SWAT library for high-throughput yeast genome tagging. Nat. Methods 15, 598–600
- Cohen, Y. and Schuldiner, M. (2011) Advanced methods for high-throughput microscopy screening of genetically modified yeast libraries. Methods Mol. Biol. 781, 127–59
- https://www.olympus-lifescience.com/en/applications/rapid-automated-detection-and-segmentation-of-glomeruli-using-self-learning-ai-technology/
- https://www.olympus-lifescience.com/en/discovery/20-examples-of-effortless-nucleus-and-cell-segmentation-using-pretrained-deep-learning-models/
University of Münster 저자:
Julian Schmidt, Sarah Weischer, Mike Wälte, Jens Wendt, Thomas Zobel, Maria Bohnert
Evident 저자:
Manoel Veiga, 응용 전문가, Evident Technology Center Europe
이 애플리케이션에 사용되는 제품
Sorry, this page is not
available in your country.