Specimen Preparation Using Synthetic Fluorophores and Immunofluorescence
Triple-Staining Adherent Cells with MitoTracker, Phalloidin (or Phallacidin), and Nuclear Dyes
The vitality and physical properties of adherent cells grown on coverslips in Petri dishes can be readily determined using a popular combination of fluorescent stains that includes one of several MitoTracker probes (for mitochondria) along with phalloidin (or phallacidin) conjugated to common low molecular weight synthetic fluorescent probes. Among the useful fluorescent markers for visualization of the filamentous actin cytoskeletal network in cells are rhodamine, fluorescein, the Alexa Fluor series, and the cyanine dyes. Counterstaining for nuclei using a variety of popular dyes follows treatment with the mitochondrial and actin probes. This protocol details a generalized procedure for staining a variety of cell types.
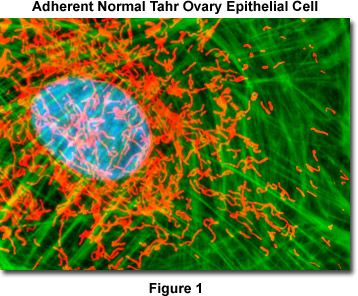
Presented in Figure 1 is a merged confocal image stack (4 optical sections) revealing the nucleus, mitochondria, and filamentous actin network in a normal Himalayan Tahr ovary (HJ1.Ov line) epithelial cell. The adherent cell culture was stained as detailed below using MitoTracker Red CMXRos in the growth medium (red), followed by phalloidin conjugated to Alexa Fluor 488, which targets polymerized actin (green). Nuclei were counterstained with Hoechst 33342. The specimen was imaged with a 100x oil immersion objective (without zoom) using the largest pinhole diameter setting on a confocal laser scanning biological microscope in combination with a 405-nanometer violet diode laser (Hoechst), a 488-nanometer argon-ion laser (Alexa Fluor 488), and a 543-nanometer helium-neon laser (MitoTracker). The images were sequentially collected in grayscale channels and subsequently pseudocolored with hues approximating the fluorescence emission spectra of the respective probes. Although this fluorophore combination produces useful images, other probes absorbing in the infrared (such as MitoTracker Deep Red 633 and the TO-PRO-3 nuclear dye) can be employed with equal success.
Reagents
- MitoTracker Medium - MitoTracker dyes are provided in vials containing 50 micrograms of the recrystallized probe. Reconstitution is done with dimethyl sulfoxide (DMSO) to make a final concentration of 1 millimolar. The amount of DMSO added depends upon the molecular weight of the dye and is listed below. After adding the DMSO, vortex the solution thoroughly and allow the mixture to sit at room temperature in the dark for 5 minutes. Repeat the vortex action,
and then centrifuge the sample to relocate scattered droplets to the bottom of the tube and dilute the appropriate aliquot. For all MitoTracker dyes, dilute 3 microliters to 10 milliliters (300 nanomolar final concentration) of growth medium to make a working solution.
- MitoTracker Red CMXRos - 94 microliters DMSO per vial.
- MitoTracker Orange CMTMRos - 117 microliters DMSO per vial.
- MitoTracker Green FM - 74 microliters DMSO per vial.
- MitoTracker Green FM - 74 microliters DMSO per vial.
- MitoTracker Red 580 - 69 microliters DMSO per vial.
- MitoTracker Deep Red 633 - 92 microliters DMSO per vial.
- MitoTracker Wash Medium - Cells are washed twice with complete medium after MitoTracker treatment, requiring 6 milliliters of wash medium at 37 degrees Celsius per Petri dish.
- Fixative - Prepare 3.7 percent paraformaldehyde dissolved in complete growth medium containing serum immediately before use. Each Petri dish will require approximately 3 milliliters of medium.
- Permeabilization Buffer - 0.2 percent Triton X-100 in PBSA (sonicate detergent buffer for 30 minutes to one hour immediately before use).
- Wash Buffer (After Permeabilization) - Use PBSA for all steps except those requiring specialized buffers for nuclear dyes.
- Blocking Buffer - 1 percent bovine serum albumen (BSA) in PBSA containing 0.05 percent Triton X-100 (add 2-3 milligrams sodium azide per 100 milliliters of blocking buffer to eliminate the growth of microorganisms).
- Phalloidin or Phallacidin Working Solution - Fluorescent phallotoxins are supplied as lyophilized solids containing 300 units of product. The contents should be dissolved in 1.5 milliliters of methanol to yield a final concentration of 200 units per milliliter. One unit is defined as the amount necessary to stain one coverslip of fixed cells and is equivalent to 5 microliters of the methanol stock solution. Prepare working dilutions of phalloidin or phallacidin by diluting the appropriate quantity of stock solution into Blocking Buffer. For example, 50 microliters of stock solution diluted to 1 milliliter with Blocking Buffer will enable staining of 10 coverslips using 100 microliters per coverslip.
- Nuclear Dye - Prepare fresh dilutions of the nuclear dye immediately prior to staining.
- Nuclear Dye Wash Buffer - Hoechst and SYTOX dyes require Hanks Balanced Salt Solution (Hanks BSS), while DAPI, as well as the monomeric and dimeric cyanine nuclear stains, can be used with PBSA.
Nuclear Counterstain Dilutions
- Hoechst (33342 and 33258) - Dilute 5 microliters of 10 milligram/milliliter stock solution in 150 milliliters of Hanks BSS (treat for 30 minutes).
- SYTOX Green and Orange - Dilute 10 microliters of concentrated stock solution (5 millimolar in dimethyl sulfoxide) in 250 milliliters of Hanks BSS (treat for 30 minutes).
- DAPI - Dilute 5 microliters of 10 milligram/milliliter stock solution in 150 milliliters of PBSA diluted 50-percent with double-distilled water (treat for 5 minutes).
Procedure
Note: MitoTracker treatment is conducted on live adherent cells. Remove medium from the growing cells and replace with prewarmed (37 degrees Celsius) medium containing serum and the MitoTracker probe at the appropriate concentration (about 350-400 nanomolar). Incubate the cells for 45 to 60 minutes in a carbon dioxide incubator.
Aspirate the MitoTracker medium from the Petri dishes, and add 3 milliliters of pre-warmed (37 degrees Celsius) medium per 60-millimeter Petri dish. Incubate the wash medium at 37 degrees for 5 minutes to allow unbound MitoTracker to diffuse into the medium. Repeat this procedure once before fixation.
Fix the adherent cells by quickly aspirating the last medium wash and adding 3.7 percent paraformaldehyde in pre-warmed (37 degrees) complete medium, including serum. Allow the cells to incubate in fixer for 15 minutes at 37 degrees Celsius.
After fixation, wash the cells with three changes of PBSA Wash Buffer (2 to 5 minutes each wash). Slowly rotate the Petri dishes containing coverslips as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Dissolve the membranes of the adherent cells with Permeabilization Buffer by treatment for 10 to 15 minutes. Slowly rotate the Petri dishes containing coverslips as they are being permeabilized on an orbital shaker at 5-10 revolutions per minute.
After permeabilization, wash the cells with three changes of PBSA-Triton Wash Buffer (5 minutes each wash). Slowly rotate the Petri dishes containing coverslips as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Block nonspecific phalloidin (or phallacidin) binding sites with Blocking Buffer for one hour. Slowly rotate the Petri dishes containing coverslips as they are being blocked on an orbital shaker at 5-10 revolutions per minute.
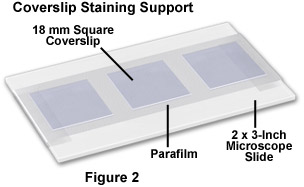
Prepare phalloidin staining supports by covering 2 × 3-inch microscope slides with Parafilm, as illustrated in Figure 2. Secure the Parafilm so that it adheres tightly and is smoothly distributed along the glass surface (no blisters). After blocking, carefully remove the coverslips from the Petri dishes and place them cell-side down on a 100 microliter drop of diluted phalloidin in blocking buffer deposited on a Parafilm-covered slide. Between 3 and 6 coverslips can be placed on a single slide. Next, place the slides in a humidity chamber covered with aluminum foil to protect the fluorophores from light (see Figure 3). Incubate the covered slides in the humidity chamber for 30 minutes at room temperature.
After phalloidin treatment, return the stained coverslips to their respective Petri dishes and wash three times with PBSA-Triton Wash Buffer (5 minutes each wash). Cover the Petri dishes with aluminum foil or an aluminum baking tray (to protect from light) and slowly rotate the cells as they are being washed on an orbital shaker at 5-10 revolutions per minute.
For DAPI and cyanine nuclear counterstains, add the diluted dye in PBSA to the Petri dish and treat the adherent cells for the recommended time: 5-10 minutes for DAPI; 15-30 minutes for cyanine dyes (protect from light with aluminum foil). When using Hoechst or SYTOX stains (30 minute incubation), first wash the cells in Hanks Balanced Salt Solution for three buffer exchanges prior to counterstaining.

Wash the counterstained cells with either PBSA or Hanks Balanced Salt Solution (depending upon the nuclear dye) for three times at 5 minutes for each wash. Protect from light with aluminum foil. Thorough washing at this stage is necessary to remove all traces of unbound phalloidin (or phallacidin).
In order to remove excess salt, wash the cells three times for 2 to 3 minutes (each wash) in distilled water. Note that this step is only necessary if the coverslips are to be air-dried overnight before mounting.
After the final distilled water washing step, carefully remove the coverslips from the Petri dish with tweezers and wipe excess water from the back and edges. Lean the coverslips on their sides against the labeled Petri dish cover and allow them to dry overnight. Protect the drying coverslips from light with an aluminum baking tray. After drying, mount the coverslips (cell-side down) on clean microscope slides using the appropriate mounting medium.
Sorry, this page is not
available in your country.



